Recently, a member of the HNPP community posted a unique website that aims to crowdsource medical information. StuffThatWorks is aiming to essentially create a databank of information for rare conditions so that people won’t be endlessly searching for treatments and understanding their symptoms. The website was created by Yael Elish, a core member of the Waze founding team. The idea was born after Yael spent a decade helping family members cope with medical conditions. She searched online endlessly and found that, time and again, other patients were the key to discovering the most effective treatments.
Here’s what it says on their website:
“Most of us dealing with a chronic condition spend hours online searching for better treatments because we feel no one has properly researched which treatments will work best for people like us. We’re right: nobody’s doing it, even for the most common or serious conditions, let alone more mundane or rare ones. It’s just too expensive to conduct large-scale patient interviews, and most medical research is done on very small groups of patients. The result? The treatments we get are far from being optimized for us.
“Just like Wikipedia, Waze or Kickstarter, when people come together for a common cause the result can be huge. We should be driving the research about our condition: we know how treatments affect us, and collectively we have more information than any organized research could ever gather. Together, we can build the world’s richest and most up-to-date database for treatment effectiveness, and use it to learn more about our condition— underlying causes, patterns in symptoms, comorbidities and which treatments work best for each of us.
“Patients dealing with chronic conditions share their experience in an organized survey. This data is then normalized, anonymized and analyzed using advanced machine learning algorithms that are programmed to look for valuable insights—for the entire condition community, for subgroups and for individuals. The insights are shared with the community, where members can comment, discuss and pose new research questions. Community members take an active role in determining the research direction. It only takes a few dozen people completing the survey to start generating useful insights. The more people join and contribute their information, the smarter and more personalized the insights become.
“Out of this experience, a small team of highly motivated people came together to create StuffThatWorks. Experts in crowdsourcing, machine learning, and medical research, StuffThatWorks is backed by three reputable VC firms, Bessemer Venture Partners, 83 North and Ofek Ventures.”
Results from an annual conference that took place late last year have revealed the extent of symptoms suffered by those with HNPP. Sufferers say that pain is the number one issue that plagues them.
On November 3rd, nearly 100 participants gathered at the Samberg Conference Center on the Massachusetts Institute of Technology (MIT) campus in Cambridge, MA, for the Hereditary Neuropathy Foundation (HNF) Patient-Centered Charcot Marie-Tooth (CMT) / Hereditary Neuropathy with Liability to Pressure Palsies (HNPP) Pain Summit. Ninety per cent of the 115 patients that took part in the study linked to the conference, indicated that their pain had a moderate to severe impact on their quality of life.
Attribution: Hereditary Neuropathy Foundation
The meeting brought together people with hereditary neuropathies and their families, caregivers, clinicians, researchers, funding agencies, sponsors, leading pain experts, and the pharma industry to provide an in-depth look at chronic pain within the CMT/HNPP community, including its impact on quality of life.
The one day conference also offered expert sessions as well as breakout session, primarily led by sufferers, with a focus on identifying gaps that are hindering patient care in neuropathic and musculoskeletal pain, diagnosis and outcome measures to support development of therapies.
Consequently, HNF partnered with the voice-powered survey platform True Reply to record patient responses to a five-question survey in their own voice. A total of 621 responses were recorded by the participants over a 30-day period.
The questions asked included:
What type of Charcot Marie-Tooth or inherited neuropathy do you have?
Do you experience disease-related or other types of pain?
Please describe your pain symptoms as descriptively as possible.
Please describe how your pain impacts your daily quality of life.
How do you currently manage your pain?
HNF founder and CEO, Allison Moore, said that the results were “enlightening”.
Moore said: “Hearing about our patients’ experience with pain in their own words was both enlightening and heartbreaking at the same time.
“Our patients are hurting badly in so many ways, and they need guidance and protocols from their healthcare providers to help manage their pain so they can go about their daily lives as pain-free as possible.”
“The voice of the CMT patient can no longer be ignored when it comes to the protocols, treatments and therapeutics that are being developed to treat this disease.”
Allison Moore, Hereditary Neuropathy Foundation CEO
Other key takeaways from the study showed that 76 per cent of patients are managing their pain on their own, with a combination of over-the-counter medications and alternative therapies, while 63 per cent described their pain as numbness, sharp, burning or stabbing.
Jose Cotto, founder and CEO of True Reply, said: “The ability of True Reply to quantitatively analyse patient responses in real-time while also giving researchers and clinicians access to qualitative data such patient voice tone, cadence and stress levels is a real game changer for Patient Reported Outcome (PRO) studies”.
Moore said she hopes that as more sufferers speak out about their experiences, the better therapies may be: “We are looking forward to using technologies like True Reply to help us tell our patients’ stories in their own words so we can address their immediate quality of life issues while waiting for desperately needed therapeutics to move through the pipeline and be approved for commercial use.”
She adds: “The voice of the CMT patient can no longer be ignored when it comes to the protocols, treatments and therapeutics that are being developed to treat this disease”.
There have been some great developments in the world of HNPP, with the most recent being a website dedicated to all research papers related to the condition.
For those who want an easier way to explore scientific publications on HNPP, behold the website HNPP Research, compiled by a fellow sufferer, who happens to be a both a software developer with experience in the medical research field.
Speaking about the website, the creator has said: “It’s not for beginners, but if you are curious to dive deeper into the scientific side I hope this will be a helpful resource.
“There are over 500 studies and I learned a lot about the underlying causes by reading the articles. You can also search on topics of interest like “pain” to see what the latest studies say.”
On the site itself, he has asked other researchers support open access journals, sharing pre-print versions with a patient support group such as HNPP and CMT / Neuropathy Support and HNPP Help.
Results from a study conducted this month by scientists at the Samsung Medical Center in South Korea, in collaboration with LabGenomics, have revealed that hereditary neuropathy with liability to pressure palsies (HNPP), is much more common than usually assumed.
Researchers estimate that one in every 5,943 Koreans have a deletion within the PMP22 gene, which is higher than worldwide estimates which stands at every 16 out of 100,000 person with the condition. It is generally believed across the board that 2-5 of 100,000 have HNPP, however, this latest research shows it is the equivalent to every 59th person out of 100,000, many of whom may present only mild symptoms. So are there more people with HNPP than we initially thought?
The team detected seven samples of those tested with deletions of genetic content affecting the 17p11.2 chromosome – which includes the PMP22 gene – and two cases of PMP22 gene duplication leading to CMT1A. They report that all samples were from unrelated families with six out of seven being female, and three (42.9%) had a family history of HNPP.
The exact prevalence of HNPP remains unclear, however, a prior study from the Republic of Ireland and in southwestern Finland reported a prevalence of 0.84 per 100,000 to 16 per 100,000. The major difference in this study is that patients with symptoms of HNPP were tested under the Irish and Finnish report, whilst South Korean researchers analysed data from newborns.
Previously reported prevalence of hereditary neuropathy with liability to pressure palsies:
Attributed to: Frequency of hereditary neuropathy with liability to pressure palsies (HNPP) due to 17p11.2 deletion in a Korean newborn population, 2018, Jong Eun Park et al.
While the study had limitations and the number of subjects may not be adequate enough to estimate the exact prevalence of the rare genetic disorder, it still provides insight regarding the baseline frequency of the chromosome deletion that commonly causes HNPP.
As the authors say: “[A] significant proportion of parents with PMP22 duplication already have CMT1A-related symptoms and may be more likely to request prenatal genetic diagnosis or preimplantation genetic diagnosis (PGD) rather than neonatal screening.
“Our data suggest that PMP22 deletion [associated with] HNPP might not be uncommon, at least in the Korean population,” they concluded.
For those who follow disability forum Team Inspire, you may have spotted Lainie from Trend-Able launching her new website with the tag line “where your disability is not an accessory”. Now this is a bit of a godsend for some women who struggle with buttons and even pulling up socks, but it seems that Lainie will be providing first-hand insight on what to use, and wear especially for those with orthotics and prosthetics.
After my work Christmas party, my legs had lost all feeling thanks to the stupidity of high heels and my own lack of commonsense. So it was only time that someone with the wits and the technological wisdom to create a wonderful site full of tips and tricks. And while it’s still a very new creation, it might be worth subscribing for any future information.
If you’re a fashionista, never fear, Lainie is here.
After several months of falling off the internet radar due to unruly fingers, I realised I may need some outside help. That’s where occupational therapists and vocational rehabilitation comes into play.
It’s easy to become confused over the role of an occupational therapist, given that it seems as if it is related solely to work-based activities. However, they cover a wide range of issues and activities that allow a person to operate relatively independently.
What is occupational therapy?
Occupational therapy is crucial in helping a person cope with the functional, vocational, and social impact of the condition. It helps a person in improving sensory motor skills through regular exercises related to it. It also teaches us to avoid exposure to certain environmental and industrial toxins that can be harmful.
The OTs also teach self care activities and patient safety issues. The therapist also teaches us to pay attention to issues which involve functions like learning how to change positions smoothly to avoid becoming numb and how to prevent falling. They can work with physiotherapists to ensure you get the best care possible.
There is a strong educational element in occupational therapy. Therapists typically teach people how to:
Prevent falls by watching out for uneven terrain and other hazards
Adjust habits, such as sitting correctly without injuring yourself
Make ergonomic changes at work and home to reduce pain and increase mobility
Find the best solutions to allow you to live independently.
Obviously, there are a lot of crossovers with vocational rehabilitation when it comes to learning to stabilise yourself. From avoiding falling at home and work, as well as correcting your posture, which can be applicable in any situation. Therefore an OT can be in charge of:
Environmental assessments – at school, work, home
Equipment recommendations
Fatigue management
Career advice
Workplace assessments
OTs are people-centred and their goal is to promote and enable independence. They will assess how well you cope with activities of daily living (ADLs), listen to your needs concerning personal care, leisure, work, study, travel and household management and advise on options for you. Their assessment may involve breaking down the activities you find hard into their component parts.
For example, if you have neuropathy you may struggle with everyday activities like getting dressed, opening food packets or holding a pen to write. Your OT will work with you to find solutions to these problems to help you remain independent. Solutions may come in the form of trying some adaptive equipment to compensate for your difficulty, or by working on activities to help maintain strength in certain muscle groups.
OTs can also make referrals for making splints for hands. People with HNPP may develop problems holding and gripping and experience some muscle wasting in their hands. A hand splint can help to keep your hand in a good position in order to minimise pain and muscle contractures.
At various stages of the condition, an OT may be able to offer expertise in areas such as:
Individualised fatigue management programmes to understand the nature of your particular fatigue within your daily life
How to more effectively prioritise and manage your time to achieve the things you want to do
Strategies to improve sleep and good quality rest
Relaxation as a coping strategy – for example as a stress or pain management technique
Ergonomic information about effective joint protection and energy conservation strategies
Hand-care techniques including provision of hand exercise programmes, fabrication of custom made hand splints to aid daily tasks, pain management and hand positioning
Adaptive equipment from small aids to major adaptations for helping you at home or in your workplace
Signposting and referring on to agencies to help with the cost of purchasing daily living aids and adaptations
Information on employment legislation and your rights within the workplace
Graded return-to-work and remaining-in-work programmes
Care assessments for direct payments or home helps
Mental health-related referrals.
In the UK, OTs work in various settings including community teams, social services and hospitals. The health professionals involved in your care, including doctors, nurses and therapists, can refer you to an occupational therapist if this is required. You may also be able to self-refer to some therapy services – so it is always worth giving your local social services a call. They will explain the correct process for your area.
Some of the adapted changes in my own home include:
A wheelie tray to be able safely carry hot items from one place to another
A food workstation – which has adapted facilities such as a place to hold objects in order to be able to cut safely, a flat grater and slicer
Adapted knives – it has better grip and position to allow you to cut object safely
Special cutlery – a bevelled fork allows you to use one hand to both cut and eat
A bath board – to be able sit safely while in a bathtub
While you may feel helpless in the face of such an uphill battle, occupational therapists go a long way in assisting you to succeed.
Useful links:
Complete Care Shop – for adaptive equipment – there is VAT relief for those with HNPP
Living Made Easy – (NHS and OT recommended) price comparison site for adaptive equipment
AbilityNet – help the lives of disabled people by helping them to use digital technology at work, at home or in education
Naidex – disability information shows like Naidex are excellent for giving you an idea of what is available, but be warned, these shows are huge. Take advantage of their Shopmobility scooters, or you’ll never last the distance
Expo Database – trade conferences around the world that showcase the latest disability equipment.
It’s a fact that most people with HNPP know, but there are suggestions that certain exercises can actually do more damage than good. From walking to some types of yoga, while most forms of physical activity are encouraged by gym instructors, our bodies tend to rebel from the norm. But can exercise actually induce HNPP, even though it’s generally thought to be an hereditary condition?
As HNPP is an autosomal dominant disorder in which the condition is inherited and there is a deletion of one of the genes associated with PMP22, a child of an affected individual is at a 50 per cent risk of being affected by the condition. This suggests that the likelihood of a person developing HNPP out of nowhere is relatively low.
What are ‘sporadic’ cases of HNPP?
There have been recent reports that state that a few types of muscle training has actually brought about sporadic symptoms of HNPP without any previous familial history of the condition. Sporadic cases due to de novo deletion accounted for 21 per cent of the investigated HNPP families, as reported by some studies. De novo mutation is an alteration in a gene that is present for the first time in one family member as a result of a mutation in a germ cell of one of the parents or in the fertilised egg itself.
According to a 2017 study carried out by researchers at the Department of Neurology and Rheumatology, Shinshu University School of Medicine, Matsumoto, Japan, this apparently was the case in a 15-year-old boy. The authors suggest that this is the first instance of an adolescent “that developed neurological symptoms during muscle training in a school baseball club activity”, the first signs in a sporadic case of HNPP. The teenager developed bilateral painless brachial plexopathy through short-term barbell training and plank exercises.
“Patients sometimes show an atypical clinical phenotype, and a diagnosis of HNPP can therefore be challenging, especially in sporadic cases as in our patient.”
“Muscle Training-induced Bilateral Brachial Plexopathy in an Adolescent with Sporadic HNPP” – Kodaira, M., et al – July 2017
While brachial plexopathy can be a common feature of HNPP, bilateral involvement is thought to be unusual because HNPP is usually associated as a mononeuropathic condition, where there is a focus in only one area of nerves.
The authors in this case say: “As his clinical and NCS [nerve conduction study] findings indicated muscle training-induced bilateral brachial plexopathy in HNPP, genetic analysis for this disorder was performed, which revealed deletion of the PMP22 gene. Patients sometimes show an atypical clinical phenotype, and a diagnosis of HNPP can therefore be challenging, especially in sporadic cases as in our patient.”
The major difference in this occasion is that not only did he suffer from a bilateral injury, he also developed a sporadic case of HNPP triggered by relatively straightforward exercises. This is unlike some of the other examples where those in military training are far more likely to develop symptoms linked to HNPP, due to the strenuous physical activity undertaken by soldiers. However, this is not the first time ‘push ups’ are seen to be the main culprit as a trigger.
In another 2017 report A Case of HNPP due to Push-up Exercise, a 17-year-old man with no familial history of the condition developed “motor and sensory disturbance of the left upper limb a few days after starting push-up exercise”. Scientists from the Department of Neurology, Hirosaki University Graduate School of Medicine, say that the patient also developed brachial plexopathy, similar to the case above.
In the abstract, they conclude that: “Genetic tests revealed a diagnosis of hereditary neuropathy with liability to pressure palsies (HNPP). HNPP should be included in the differential diagnosis for neuropathy due to slight exercise or nerve compression even when familial history is negative.”
Researchers from the Division of Neurology and the Department of Pathology, University of Missouri School of Medicine, Columbia, USA, found a case in 2004 akin to the above studies. A 21-year-old in good health began to develop symptoms of HNPP on her first day of military training. She began to show symptoms of severe pain, weakness, and atrophy in her right shoulder, foot and hands. Her mother and her family had no history of neuromuscular disease. She did not know her father or his family history.
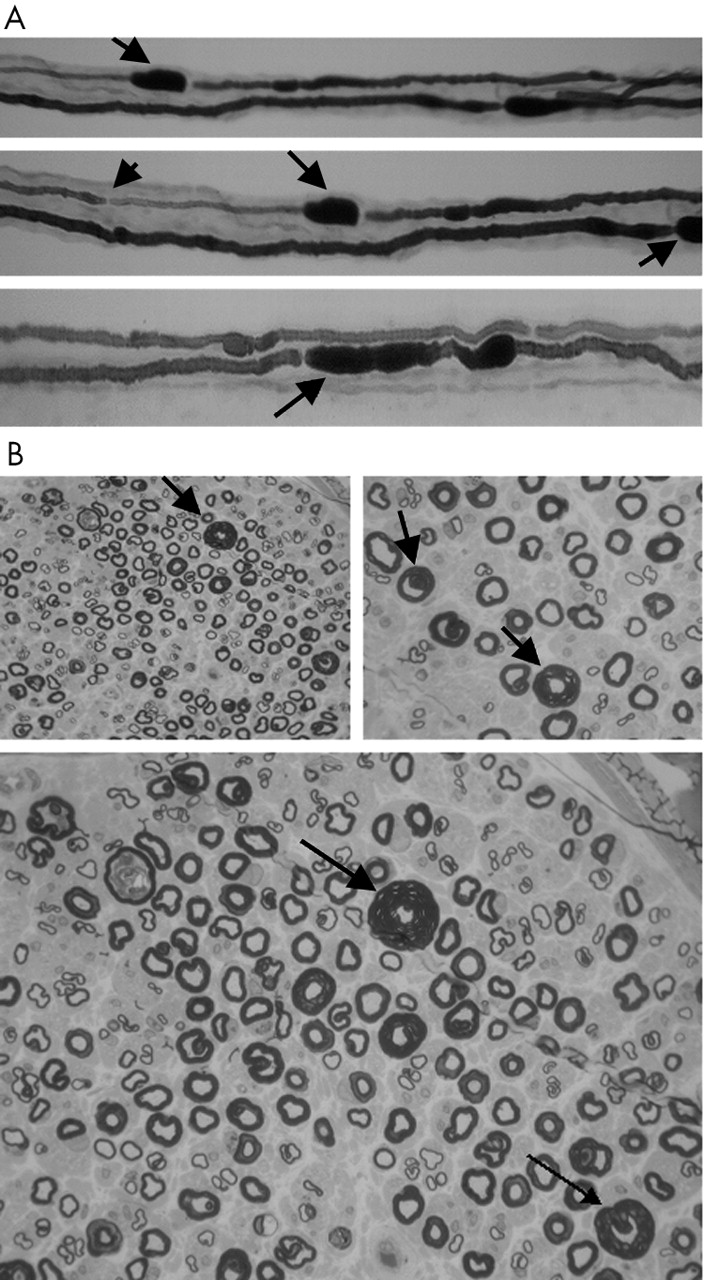
(A) Teased fibre preparation of the right sural nerve showing focal sausage shaped enlargements of the myelin sheath (tomaculae) (indicated by solid arrows) and thinly remyelinated internodes (indicated by arrow head). (B) Epon section of sural nerve demonstrating marked variation in myelin thickness and several large hypermyelinated axons (indicated by solid arrows).
They say: “While the presentation and severity of this patient’s condition may relate to a specific unknown genetic profile with very low PMP-22 mRNA levels, Schenone et al, in finding correlations between reduced PMP-22 mRNA levels and disease severity, also suggested that extrinsic factors—for example, level of physical activity, may be important in determining phenotypic features.
“This appears to be true of our patient—that she was neurologically normal, then developed symptoms on the first day of military physical training with progression as she continued the training over a three week period, suggests that disease severity and focal axonal damage were related to these intense activities.”
The authors add: “Additionally, as it has recently been recognised that sporadic cases of HNPP are common, either because of de novo mutations or asymptomatic carriers, her lack of family history did not preclude this diagnosis.”
How likely is it that a case of HNPP is sporadic?
It’s important to note that HNPP may not be easily traced in other family members, which means while it may appear sporadic, it could just have been overlooked. A 2013 case report reiterates this view, with authors saying that approximately one-third of deletion carriers are unambiguously detected on the basis of “electrophysiological criteria and confirmed by genetic analysis are asymptomatic and do not display significant signs at clinical examination.”
Researchers from Department of Paediatrics, Hospital de Guimarães, Portugal, say: “Thus, the family history is often uninformative, and a significant proportion of probands may be considered as apparently sporadic cases. However, a close questioning and examination of the relatives provided evidence for autosomal dominant inheritance in families that were originally stated by the probands to be normal. Therefore, HNPP can easily be overlooked in those cases in which familial involvement is not recognised unless intensive ascertainment techniques are used.”
Electrophysiologic studies are said to be “suggestive” and not “sufficient for diagnosis” which may be why it may be imperceptible to locate.
How exercise can trigger symptoms
The more frequent representations are in those who already have familial histories of HNPP, or in those who have already been diagnosed. In one such case in 2005, hiking and athletic training brought on the symptoms. A 10-year-old girl, who suffered from acute, recurrent monoplegic episodes affecting both the sciatic nerves and the left brachial
plexus since the age of 7, showed quite a lot extensive symptoms from relatively low impact exercise.
Authors from the Department of Paediatrics, Yokohama City University Medical Centre, Japan, say: “This school child having HNPP is considered to be susceptible to the influence of abundant physical training, rather than minor trauma or compression at sites of entrapment of peripheral nerves.” However, this is hardly surprising given the nature of the condition. What’s more unusual are the situations where HNPP is not present beforehand or without familial background.
It goes without saying, exercise can cause many types of injuries even for those without HNPP depending on how extreme it may be. And as we have seen above, there have been at least several cases of ‘sporadic’ HNPP, which develops without any prior history of symptoms or familial connection.
However, it may be worth noting that without the correct tests to detect hereditary links, these may be incorrectly termed as ‘sporadic’. That being said, there have been established reports that suggest at least 21 per cent of HNPP cases are de novo mutations, so it may not be out of the realms of possibility that certain types of exercise can induce symptoms of HNPP.
There can be a lot of worry and anxiety that arises during pregnancy, one of which includes what to expect, especially with HNPP. Many can have a more or less seamless experience while others find that their symptoms are exacerbated during this time. So what is the ‘norm’ of HNPP during pregnancy?
“In my 30’s during pregnancy, I had sciatica because I had enormous babies resting on my spine. Bladder too, but peeing my pants when I sneezed or laughed – seemed like something that just happens to pregnant women.”
First of all, HNPP does not affect the fetus or the pregnancy itself thankfully. However, during pregnancy, symptoms that manifest due to HNPP such as palsies, sciatica, or pain in the lumbar region, may be heightened as a result of added pressure on the body.
Disclaimer: Please ask your medical practitioner for more information. This article is based on various research, journals and testimonies.
How does HNPP manifest during pregnancy?
According to Dr. Rakesh B Vadhera, an obstetrics anaesthesiology consultant and professor at the University of Texas, alongside Dr. Michelle Simon, a paediatrician and neuropathology expert, peripheral entrapment neuropathies are common during pregnancy and may lead to “severe discomfort”.
Writing in the book Maternal Medicine published in 2015, Dr. Vadhera and Dr. Simon state: “Pregnancy itself may predispose patients to some of these entrapment neuropathies, which are mostly benign in their evolution and prognosis and will resolve spontaneously in the postpartum period.” This appears to be good news for expecting mothers concerned that the symptoms may not disappear after the birth of the child. For all that however, there have been cases of symptoms lingering postpartum.
They add: “Delivery may predispose patients to compression or stretching of some nerves and plexuses that may precipitate symptoms. Prompt clinical evaluation and, when necessary, an electrophysiologic evaluation may aid in the diagnosis and subsequent management.” As briefly mentioned in the article Is surgery worth it with HNPP?, it’s vital to let your medical team know how to make you comfortable during this time as well as through labour, to avoid further nerve-related damage. This is addressed in more detail below.
What symptoms to expect when you’re expecting
In some extreme cases of Charcot Marie-tooth-related disorders, the obstetricians above say pregnancy can affect respiratory muscles and thoracic vertebral anatomy, “impacting patient respiratory function during pregnancy and affecting delivery and anesthetic care”. But this may be evident during the third trimester when there is added strain on the body, and therefore you may have enough warning to consult a health professional beforehand.
Author Dr. Pierre Bouche, based in the Department of Clinical Neurophysiology, Salpêtrière Hospital, Paris, France, says that in some neuromuscular disorders, carpel tunnel syndrome (CTS) could also manifest during pregnancy.
In the edition Peripheral Nerve Disorders as part of the Handbook of Clinical Neurology, Dr. Bouche states: “[Carpal tunnel syndrome] can develop at any time in pregnancy, but it is most frequent during the third trimester and may be due to fluid retention exerting pressure on the median nerve.” However, this can vary from person to person depending on how sensitive the nerves are around the wrist and upper arm.
Other areas that may be affected can also differ. Authors of the medical reference guide Obstetric Anesthesia and Uncommon Disorders, 2008, reiterate that HNPP may exacerbate neuropathies associated with pregnancy and delivery. They say HNPP symptoms such as “lumbosacral plexus, femoral, lateral femoral cutaneous, obturator or peroneal nerve palsies” may be aggravated during this time.
But that’s just some of the ways the symptoms may manifest. There are some mothers featured in the Facebook HNPP groups, who have spoken about pain in the ribs, loss of functionality in the legs, arm and leg aches, and the list goes on. On the other hand, there are others who faced symptoms no worse than pre-pregnancy.
How to prepare for labour and delivery
Similar to the diverse responses on how mothers are affected during pregnancy, the same is apparent with the delivery itself. Some mothers elect to have a natural birth, while others require or request cesareans. Using gas, on the other hand, may pose a risk as it is considered a neurotoxin. There have been reports of “heightened pain” with gas according to some users in the HNPP support networks.
“I was 33 when I had the epidural – which triggered my chronic neuropathic pain. The majority of my pain, travelling along the entire right side of my body. Strongest in all the places I had experienced pain during my life. It was like it was the “Red Button” got pushed and a bomb exploded in my Central Nervous System.”
Consult with a neurologist and anaesthesiologist in the antenatal period
Assess neurological status antepartum
Avoid prolonged immobilisation in labour
Avoid instrumental delivery
Avoid dense epidural blockage
Consider operative delivery if a pressure palsy develops during labour
If a cesarean section is selected, HNPP.org gives the following advice to the surgical team:
Position arms out to sides. An angle of less than a 90 degree angle will help to alleviate stretch on the brachial plexus (shoulder area).
Move arms (supinate/pronate) every 15 minutes while under general anaesthesia.
Pad arms and legs/feet in stirrups. As a general rule: pad everything. The need to pad arms and legs is dependent upon the individual patient (frequency and severity of palsies). One inch foam or similar type material is usually sufficient.
If possible avoid leaning against the patient, especially against the arms and legs.
Tape endotracheal tube more centrally so that the tube is fully supported by the tape and not at all by the mouth. Tape other tubing in a similar manner as appropriate. Consider positioning while awake.
In order not to mask any developing neuropathy, anything but the mildest block for postoperative pain should be avoided.
Both Dr. Lepski and Dr. Alderson say that the “Labour progressed uneventfully and there were no neurological sequelae following delivery”.
Be watchful for patient position that contributes to nerve compression, particularly with neuraxial blockade.
Avoid prolonged use of the lithotomy position; regularly reduce hip flexion and abduction.
Avoid prolonged positioning that may cause compression of the sciatic or peroneal nerve.
Place the hip wedge under the bony pelvis rather than the buttock.
Use low-dose local anaesthetic / opioid combinations during labour to minimise numbness and allow maximum mobility.
Encourage the parturient to change position regularly.
Ensure that those caring for women receiving low-dose local anaesthetic / opioid combinations understand that numbness or weakness may be signs of nerve compression; such symptoms should prompt and immediate change of position.
A report by French researchers S. Berdai and D. Benhamou from the Department of Anaesthesia and Resuscitation, Bicêtre Hospital, Le Kremlin-Bicêtre, suggests that it is possible to have an epidural as well as spinal anaesthesia during labour. In the report Regional Anaesthesia for Labor and Delivery in a Parturient with Neuropathy with Liability to Pressure Palsy, a woman had two cesarean sections, one with an epidural that resulted in no “neurologic complaints in the postpartum periods”.
They say: “For the first delivery, epidural analgesia was performed for labour pain control but a caesarean section was necessary because of failure to progress (0.0625% bupivacaine with 0,2 μg/ml sufentanil for labour then 2% lidocaine with adrenaline for surgery).
“Two years later, the patient was again seen for a preanaesthetic visit because elective Caesarean section was planned. Spinal anaesthesia using hyperbaric bupivacaine and sufentanil was used. Both deliveries were uneventful”. Uneventful being the operative word.
It is essential to get the right advice while pregnant as well as during childbirth itself, and also on how to manage any symptoms that appear postpartum. Creating a birthing plan will therefore be necessary to avoid any extra issues. That being said, symptoms fluctuate from person to person, which means you may be fortunate enough to have hardly any bumps in the road.
When your face unexpectedly becomes numb, or begins to spasm, many HNPP sufferers do wonder what godforsaken cause could be behind it. Yet, it isn’t as uncommon as many may believe. Health professionals say that facial issues tend not to be associated with the condition, hence multitudes have been diagnosed with Bell’s Palsy instead.
What is Bell’s Palsy?
Bell’s palsy, or idiopathic facial paralysis (IFP), is the most common cause of unilateral, lower motor facial palsy. It’s origins remain uncertain. However, the first familial occurrence was found in 1887, hence hereditary factors have been considered to play a role in the etiology of the disease. It is believed to occur when the nerve that controls the muscles in your face becomes compressed and IFP is generally linked to inflammation or viral infections.
“In the last year and a half I have been dealing with one sided facial pain and numbness. It has evolved into a burning that sequentially involves the tongue, then lips, then cheek, then eye, then ear and finally throat all on the left side. It waxes and wanes and seems to do so based on my stress levels. I have had CT and MRI, normal; as well as a battery of blood tests, normal; seen a neurologist who said I’m fine (ahem!) and an ear nose and throat doc who says I have non-motor Bell’s Palsy (my research indicates there is no such thing).”
It’s obvious that there could be secondary condition causing facial issues, but several members of various HNPP groups have spoken about pain, weakness numbness, and spasms associated with the head and face.
Scientists have been studying the link between IFP and HNPP in order to get a better understanding of both causes. A letter in the Journal of Clinical Neuroscience in 2013 shows how the two conditions are not connected. Researchers at the Department of Neurology, Eginition Hospital, in Athens, Greece, hypothesised that a handful of participants in a study for Bell’s Palsy could have the same mutation for HNPP.
They say: “There are a few case reports of patients belonging to these subcategories of Bell’s palsy, on whom the characteristic deletion of a 1.5-Mb region on chromosome 17q11.2-12 which includes the peripheral myelin protein 22 (PMP22) gene, was detected.”
Out of a 145 unrelated Greek patients with Bell’s palsy, 28 patients with recurrent facial palsy and 18 patients with familial facial palsy were tested for a deletion of the PMP22 gene. However, none of the participants had this mutation.
“These cases could be part of a diverse spectrum of miscellaneous disorders including HNPP.”
“Is there a common genetic background?” Karadima, G. et al, 2013
They conclude: “Bell’s palsy seems to have a different etiology than HNPP. The same applies to familial or recurrent Bell’s palsy. A molecular genetic investigation for HNPP seems to be indicated in cases of recurrent or familial facial palsy accompanied by peripheral nervous system damage or exhibiting a family history of peripheral neuropathy. These cases could be part of a diverse spectrum of miscellaneous disorders including HNPP.”
This is reiterated in the 2009 case report Familial Recurrent Bell’s palsy conducted by researchers from the Department of Neurology, Zhejiang University, Hangzhou, China. Three families in which eight patients had a total of 12 episodes of typical Bell’s palsy were recorded in a bid to find the etiology of the condition.
In spite of this, the researchers infer: “Recurrent facial palsy can occur in neurological disorders such as Melkersson-Rosenthal syndrome (MRS), Moebius syndrome, Charcot-Marie-Tooth disease and hereditary neuropathy with liability to pressure palsy (HNPP). These conditions however, have additional features that distinguish them from idiopathic familials Bell’s palsy. None of our patients have any symptoms indicative of such diseases.”
So what’s the reasons behind facial numbness?
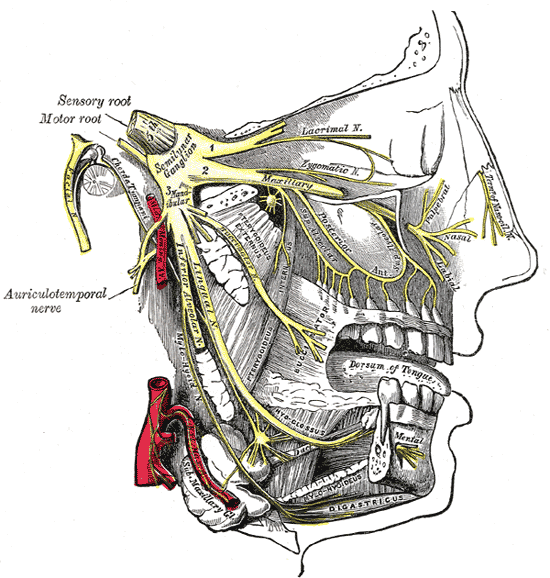
There are several possible causes of facial numbness, also known as hypesthesia. Most of these causes can be traced to a problem in or affecting the trigeminal nerve.
It is one of twelve cranial nerves and is one of the most widely distributed nerves in the head. The cranial nerves can be categorised as two main nerve types: those that control motor responses such as blinking, chewing, or eye muscle movement, and those that respond to the sensations of taste, smell, hearing, and touch.
The trigeminal nerve has three branches, which controls both the sense of touch in areas in the face as well as the motor function associated with chewing. Damage to this nerve could, therefore, make chewing difficult, if not impossible. Some sufferers of face numbness also experience numb lips. Or it could create either a ‘pins and needles’ sensation or a loss of feeling in parts of the face. Of the twelve facial nerves, it is usually considered number five. Other parts affected include:
Olfactory nerve (number 1) – relays the sense of smell to the brain.
Oculomotor nerve (number 3) – controls the external muscles of the eye.
Facial nerve (number 7) – controls the muscles used in facial expressions and should not be confused with the trigeminal nerve, despite its name. It does not relay a sense of touch.
Auditory nerve (number 8) – controls balance and hearing.
While most of these are connected with the central nervous system, and HNPP is yet to have established links to the CNS, there have been cases where some with the condition have had issues with this particular nerve.
According to a 2015 study carried out by Japanese researchers from Department of Neurology, Osaka Red Cross Hospital, Osaka, there were two cases with cranial involvement without progressive muscular atrophy (PMA). They state: “a 40-year-old female case of HNPP with the involvement of the trigeminal, facial and hypoglossal nerves, and a case of 7-year-old boy having a homozygous deletion of PMP22, who had the LMN [lower motor neuron] impairment in the cranial nerves of VII and III, sensory disturbance in extremities.”
Like many other publications, the researchers state that because of limited studies, “additional investigations are warranted to better understand PMP22 regulation in the CNS and the peripheral nervous system”.
The rarity of such finding is highlighted in a Brazilian study from the Department of Neuroscience at the University of São Paulo. In the 2016 study Clinical and Neurophysiological Features of HNPP, 39 patients were reviewed for neurological symptoms while 33 were given nerve conduction tests. Only one presented cranial nerve related symptoms in terms of “involvement of the trigeminal nerve and other one an episodic involvement of the eyelid branch of the oculomotor nerve”.
They go on to restate: “Cranial nerve involvement was rare in our population […] It seems that this is the pattern in most studies Interestingly, we have previously described a HNPP patient that developed dysphagia. Other rare manifestations in our patients were pes cavus and nerve thickening, as seems to be the case in most studies.”
In the 2006 book Differential Diagnosis in Neurology by Robert J. Schwartzman, MD, the Professor of Neurology documents daily morning reports with neurology residents and the examination of patients in front of colleagues over the last 30 years. The Emeritus Professor of Neurology at the Drexel University College of Medicine in Philadelphia, recounts that “facial nerve involvement occurs rarely” with HNPP.
Therefore, it’s important to realise the symptoms instead of the condition as knowing that it is HNPP, or in this case, may not be HNPP-related, does not change the fact that these symptoms are manifesting.
For many with facial issues, including those suffering from HNPP, it’s vital to get it checked by a medical professional as it could be associated with another underlying or even acute disorder. In many situations, doctors may dismiss it as HNPP and then resort to medication associated with the inherited disorder, because they aren’t certain of what it may be, which is where your persistence to get the correct treatment will be absolutely key.