
One of the first responses that seems to appear on search engines about Hereditary Neuropathy with Liability to Pressure Palsies is its relation to the inherited condition Charcot Marie-tooth syndrome (group of disorders). While CMT1A – a subtype of the CMT – is thought to be the most common inherited neuropathic condition, little research is still available to its ‘sister’ condition HNPP. So how do the conditions diverge and converge?
The irony of the conditions being similar in nature, is the fact HNPP is the genetic opposite of CMT1A. HNPP is associated with a deletion on the same chromosomal site where CMT1A (the most common type of CMT) has been found to have a duplication, and it contains an important myelin gene, peripheral myelin protein-22 gene (PMP22).
The chromosome in question is chromosome 17, where CMT1A has an extra copy of PMP22, and HNPP results from the loss of a copy of PMP22. The PMP22 gene encodes for peripheral myelin protein, and disruption of this gene leads to a dysfunctional myelin sheath on nerves.
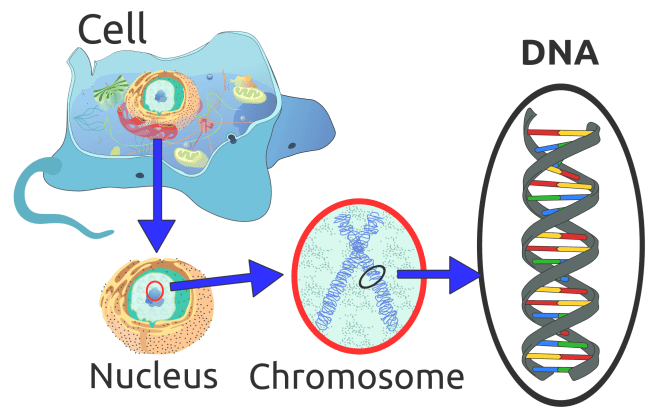
What is a chromosome?
Going back a step and remembering the old science lessons from school, a cell’s nucleus contains chromosomes – rod shaped bodies found in the nucleus of cells that contain genetic information in the form of DNA.
DNA (deoxyribose nucleic acid) is made up of short sections of genes – inherited from parents, and makes up DNA which controls part of a cell’s chemistry, particularly protein production. Each gene codes for a specific protein by specifying the order in which amino acids must be joined together.

In total, humans normally have 46 chromosomes in each cell, divided into 23 pairs. Two copies of chromosome 17, one copy inherited from each parent, form one of the pairs. In one of the arms of the chromosome 17 (if you think about it as an ‘X’ type shape), specifically the ‘P-arm’ lies the gene PMP22 labelled as 17p11.2, which is the area where it resides. The genetic defect in most HNPP patients is a 1.5 Mb deletion (used to describe the length of a DNA/RNA molecule) on this chromosome containing the PMP22 gene.
HNPP is usually caused by an autosomal dominant gene, which means one parent must be affected. While there should be two copies of the PMP22 gene, there usually is only one or some form of mutation, unlike with CMT1A which tends to be a duplication of the gene. As a result, HNPP and CMT tend to be lumped together under one category and while there may be an overlap, there are other forms of CMT and HNPP that do not follow this rule.
The result of this is that the symptoms of each condition resemble one another because they both end up becoming a form of demyelinating neuropathy.
Rare forms of HNPP
Point mutations in the PMP22 gene are a rare cause of HNPP. In one case, a novel PMP22 splice site mutation – a genetic mutation that inserts, deletes or changes a number of nucleotides in the specific site – was reported in an HNPP family. Nucleotides are organic molecules that form DNA and RNA (ribonucleic acid, which is an important molecule with long chains of nucleotides).
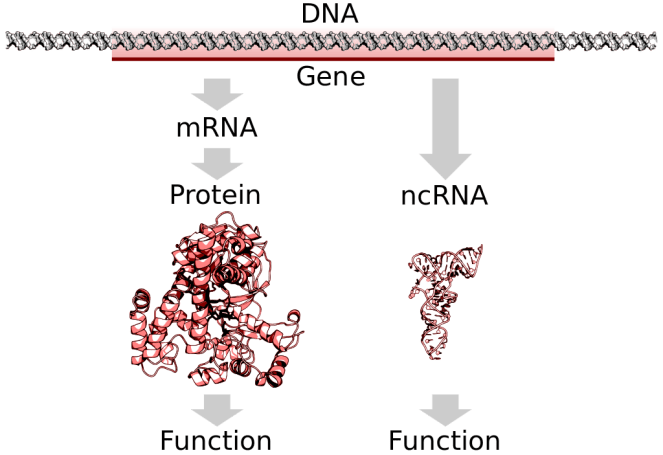
According to a 2006 study, a mutation at nucleotide c.179+1 was found in the PMP22 gene. This mutation causes the synthesis of an abnormal mRNA. Messenger RNA (mRNA), are molecules in cells that carries codes from the DNA in the nucleus to the sites of protein synthesis in the cytoplasm (the ribosomes).
Point mutations of PMP22 gene cause a wide variety of demyelinating neuropathies including HNPP, Charcot‐Marie‐Tooth disease type 1A (CMT1A), Dejerine‐Sottas syndrome (DSS), and congenital hypomyelination (CH). Authors of the 2006 study say: “To date, 11 mutations have been reported in HNPP patients and all of them are likely to cause a loss of function of the protein.” An earlier 2003 report suggested there were “fewer than 10 point mutations of the PMP22 gene” associated with HNPP.
Rare forms of CMT
CMT1B
CMT1B is the second most common subtype of CMT1. CMT1B is caused by a defect within the MPZ gene, which lies on chromosome 1. The MPZ gene produces myelin protein zero, and disruption of this gene also causes deficits within the myelin sheath. CMT1B patients have onset and symptoms similar to those of CMT1A patients, although there is a wide range of variability within CMT1B. As discussed in the article When HNPP ‘Causes Breathing Problems’, the MPZ gene is known to be associated with respiratory issues which is less common with HNPP.
CMT1E
Similar to point mutations in HNPP, instead of having a duplication of the normal PMP22 gene, CMT1E patients harbour different genetic abnormalities in the PMP22 gene.
CMTX
CMTX is caused by mutations in the gene for connexin 32, which normally codes for a protein located in myelin, the insulating sheath that surrounds nerve fibres. It has many of the same symptoms of CMT1 and CMT2, including muscle weakness and atrophy, and changes in sensation, mostly in the feet, lower legs, hands and forearms. However, because of its linkage to the X chromosome, CMTX often affects males more severely than females.
CMT2
CMT Type 2 (CMT2) is a subtype of CMT that is similar to CMT1 but is less common. It is typically autosomal dominant, but in some cases can be recessive. CMT2 is caused by direct damage to the nerve axon itself in comparison to CMT1 which results from damage to the myelin sheath insulating the axon. CMT2 is commonly referred to as “axonal” CMT.
CMT2A is the most common subtype of CMT2 and is caused by defects in the MFN2 gene. The MFN2 gene encodes for Mitofusin 2, which is a protein involved in the fusion of cellular mitochondria. Other more rare forms of CMT2 and their gene defects include:
- CMT2B is caused by defects in the RAB7 gene.
- CMT2C is caused by defects in the TRPV4 gene.
- CMT2D is caused by defects in the GARS gene.
- CMT2E is caused by defects in the NEFL gene.
CMT4
CMT4 is a rare subtype of CMT that is inherited in an autosomal recessive pattern. Generally, cases of CMT4 present with more severe symptoms compared to CMT1 or CMT2. In general, CMT4 is caused by defects in the myelin sheath which insulates the axon. However, other variations include:
- CMT4A is caused by defects in the GDAP1 gene.
- CMT4B is caused by defects in the genes MTMR2 (CMT4B1), or MTMR13 (CMT4B2).
- CMT4C is caused by defects in the SH3TC2 gene.
- CMT4D is caused by defects in the NDRG1 gene.
- CMT4E is caused by defects in the EGR2 gene.
- CMT4F is caused by defects in the PRX gene.
- CMT4H is caused by defects in the FDG4 gene.
- CMT4J is caused by defects in the FIG4 gene.
Source: CMT USA
Chronic Inflammatory Demyelinating Polyneuropathy
Asked about the similarities of Chronic inflammatory demyelinating polyneuropathy with HNPP and CMT, the symptoms that manifest seem to overlap with HNPP and CMT. However, the major difference is the fact that it is not an inherited condition, but a neurological disorder that causes progressive weakness and impaired sensory function in the legs and arms. No clear genetic predisposition or other predisposing factors for CIDP have been identified.
CIDP is thought to be caused by the immune system mistakenly attacking and damaging the myelin sheath of the peripheral nerves hence the symptoms may appear to be similar to HNPP and CMT. It is said that many people with CMT are initially diagnosed with CIDP due to these symptoms.
After that short science lesson, it’s clear to see how easily one condition can be mistakenly diagnosed for another. The symptoms tend to overlap with one another because the end result can include damage to the myelin sheath and neuropathic tendencies. As a result, it requires careful analysis by health professionals, and the necessity that all essential medical tests are undertaken to get the correct answers.
Read: Why do some HNPP results appear negative?
Read: HNPP, belief and the impact of misdiagnosis

I was diagnosed with CMT TYPE 1a. But am curious to know if. Possibly I have both CMT n CIDP because of the agressive progression n also. The inflammation. Is daily. Both. Myleath n anoxal. , are deteriorating. I was working. Full time. 2.5 years ago n now I’m. Bed ridden. From. The pain. It continues to progress. With really no treatment. It sometimes feels hopeless
How can. They diagnose. CIDP. The CMT type 1a was diagnosed by gene tic study finding 2 rare variants.
Thank you. For any answers.
Also is there any Doctors. You can recommend. In the. Stockton Calif area. I can see.
LikeLiked by 1 person
You could see Dr. Bhatia in Fresno. He is a very good neurologist that handles my CIDP.
LikeLiked by 1 person